¿Qué es la anemia falciforme?

Genética
La Enfermedad de Células Falciformes (ECF), también denominada anemia falciforme, es una enfermedad hereditaria de la sangre
La Enfermedad de Células Falciformes (ECF) es una enfermedad hereditaria de la sangre, es decir, se transmite de generación en generación. Por esta razón es muy importante tener en cuenta la diferencia que hay entre padecer la ECF o tener el rasgo falciforme.
Cada persona hereda de sus padres una copia del mismo gen, uno que proviene de la madre y otra del padre. Cuando una persona padece la ECF significa que ha heredado ambos genes de la hemoglobina falciforme (HbS) por parte de los padres y presenta todos los síntomas relacionados con esta enfermedad. Por otro lado, una persona con el rasgo falciforme o portadora solo contiene una copia del gen HbS, y generalmente se considera sana. En este caso el mayor riesgo recae en la transferencia del gen de la hemoglobina falciforme a la descendencia.
A continuación, se presenta una sesión formativa dirigida especialmente a personas con rasgo falciforme, que explica lo que significa tener el rasgo, y proporcionara recursos y recomendaciones para la prevención de la ECF.
Nuestras células contienen diferentes genes que heredamos en pares de nuestros padres. Cada par tiene una función única, como la determinación del color de los ojos o el tono de la piel. Hay un conjunto de genes que determina el funcionamiento correcto de los glóbulos rojos, lo cual no es una característica apreciable a simple vista. Estos incluyen también los genes que codifican la hemoglobina, una proteína de los glóbulos rojos responsable del transporte de oxígeno en el organismo. Se hereda un gen de la hemoglobina (Hb) de cada progenitor.
Las personas portadoras del rasgo de células falciformes tienen un gen de hemoglobina normal (HbA) y un gen de hemoglobina falciforme (HbS). El rasgo de células falciformes no produce la enfermedad; posiblemente puede causar síntomas, pero los casos son infrecuentes.
El riesgo de transmitir la ECF varía dependiendo de si cada padre tiene el rasgo o si tiene la enfermedad. Por ejemplo, cuando ambos padres tienen el rasgo de células falciformes, hay un 50 % de posibilidades de que su hijo/a también lo tenga, un 25 % de posibilidades de que el hijo/a no lo tenga y un 25 % de probabilidades de que el hijo/a herede la ECF.
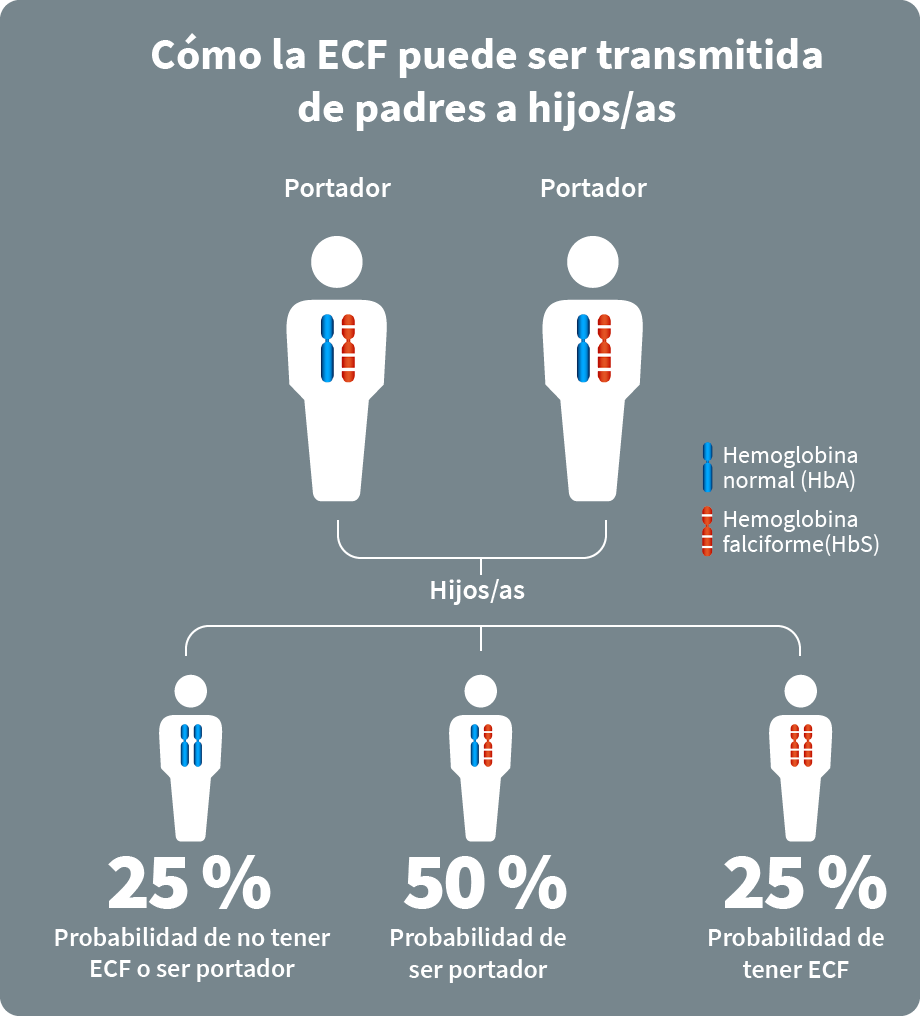

La ECF en realidad hace referencia a un grupo de diferentes tipos de trastornos sanguíneos causados por la hemoglobina falciforme. Cuando ambos padres tienen HbS, su hijo/a puede tener HbSS, que es el tipo más frecuente de ECF en todo el mundo. Otros tipos son HbSC, HbSβ-talasemia, HbSD, HbSE y HbS0. El tipo concreto de enfermedad que padece una persona depende del tipo de hemoglobina, además de la HbS, que reciba de sus padres. Cualquier combinación de HbS transmitida con otro gen de hemoglobina anormal, como HbC o Hbβ-talasemia, puede causar que un niño nazca con ECF.
Impacto en los glóbulos rojos
La ECF hace que los glóbulos rojos sean menos estables
El gen de la HbS hace que los glóbulos rojos se vuelvan rígidos y tengan forma de hoz (llamados falciformes), lo que afecta a su funcionalidad.
Estos glóbulos rojos falciformes se destruyen más deprisa que los glóbulos rojos normales y al organismo le resulta difícil volver a producir las células sanguíneas que necesita con la rapidez suficiente.
Esto lleva a lo que se llama <i>anemia</i>, una condición que produce debilidad y cansancio en la persona afectada por ECF.
La forma de disco de los glóbulos rojos les permite penetrar en los vasos sanguíneos pequeños sin sufrir daños y transportar oxígeno a través del organismo.

La forma rígida en hoz hace que los glóbulos rojos sean inestables, menos eficaces para transportar oxígeno y dificulta su paso por los vasos sanguíneos pequeños.

La destrucción de los glóbulos rojos (hemólisis) es normal, pero en las personas con ECF ocurre con mayor frecuencia.
Un glóbulo rojo con forma redondeada puede vivir en el torrente sanguíneo hasta 4 meses. Sin embargo, un glóbulo rojo falciforme puede destruirse en menos de 3 semanas. A este ritmo, el hígado no es capaz de filtrarlos a tiempo provocando el color amarillo que se observa en los ojos de los pacientes con ECF.

El organismo intentará producir nuevos glóbulos rojos para reemplazar a los que se destruyen, pero es difícil producir las células sanguíneas que necesita a la velocidad suficiente. Esto crea un problema llamado anemia, condición que produce debilidad y cansancio prolongado. Si la anemia es grave, pueden ser necesarias transfusiones de sangre.
¿Qué causa las crisis de dolor en la ECF?
La ECF va más allá de los glóbulos rojos y afecta de manera continua y silenciosa a los vasos sanguíneos y a otras células de la sangre, como los glóbulos blancos y las plaquetas, causando la adhesión de las células sanguíneas a las paredes de los vasos sanguíneos y entre sí.
La vasooclusión y las crisis vasooclusivas (CVO) continuas (a menudo silenciosas), están asociadas a un mayor riesgo de daño a los órganos, que puede llevar a fallo multiorgánico y posiblemente a la muerte.
¿Qué es una crisis de dolor?
Cuando se produce la vasooclusión, hay una falta de oxígeno en algunos tejidos y/u órganos. La falta de oxígeno puede provocar episodios de dolor, también denominados crisis de dolor, que pueden ser muy intensos y requerir atención médica. A menudo, las personas sufren las crisis de dolor en casa, sin buscar ayuda ni apoyo médico. Las crisis de dolor pueden ser frecuentes y con el tiempo pueden empeorar la enfermedad.
Por eso, es importante hacer un correcto seguimiento de cada crisis experimentada e informar a su médico regularmente.
Mitos y realidades sobre la ECF
A medida que aumenta el conocimiento sobre la ECF en todo el mundo, es importante desmitificar ciertas creencias entorno a esta enfermedad. Los siguientes mitos son frecuentes y pueden afectar a las personas que padecen la enfermedad, sus familiares y cuidadores, amigos y profesionales sanitarios.
Realidad: Si bien la ECF afecta principalmente a personas de ascendencia africana, también afecta a personas de diferentes regiones del mundo.
Realidad: La ECF es una enfermedad de la sangre hereditaria que no se contagia en absoluto. Solo se puede transmitir de padres a hijos biológicos.
Realidad: Aunque algunas crisis de dolor pueden ser de corta duración, las personas que tienen ECF, la tienen desde su nacimiento y la tendrán toda su vida.
Realidad: En realidad, existen varios tipos de ECF, que incluyen, entre otros, HbSC, HbSβ-talasemia y HbSS, que es la forma más frecuente de la afección.
Realidad: Hay más aspectos implicados además de los glóbulos rojos. Las crisis de dolor son en realidad el resultado del modo en que la ECF afecta a otros componentes del sistema sanguíneo del organismo, no solo a los glóbulos rojos.


En un centro de referencia pueden ayudarle a resolverlas, encuentre en el mapa el más próximo a su ubicación o localidad.
- Adam SS, et al. Depression, quality of life, and medical resource utilization in sickle cell disease. Blood Adv. 2017;1(23):1983-92.
- Adeyoju AB, et al. Priapism in sickle-cell disease; incidence, risk factors and complications – an international multicentre study. BJU Int. 2002;90(9):898-902.
- Anie KA, et al. Psychosocial impact of sickle cell disorder: perspectives from a Nigerian setting. Global Health. 2010;6:2.
- Ashley-Koch A, et al. Sickle hemoglobin (HbS) allele and sickle cell disease: a HuGE review. Am J Epidemiol. 2000;151(9):839-45.
- Ballas SK, et al. Sickle cell pain: a critical reappraisal. Blood. 2012;120(18):3647-56.
- Brandow AM, et al. Postdischarge pain, functional limitations and impact on caregivers of children with sickle cell disease treated for painful events. Br J Haematol. 2009;144(5):782-8.
- Cancho EJB, et al. Update of the Spanish Registry of Haemoglobinopathies in Children and Adults. Med Clin (Barc). 2020; doi: 10.1016/j.medcli.2019.10.011. [Epub ahead of print].
- Castro IPS, Viana MB. Cognitive profile of children with sickle cell anemia compared to healthy controls. J Pediatr (Rio J). 2019;95(4):451-7.
- Cela E, et al. National registry of hemoglobinopathies in Spain (REPHem). Pediatr Blood Cancer. 2017;64(7).
- Centers for Disease Control and Prevention (CDC). Sickle Cell Disease [Internet]. 2019. Disponible en: https://www.cdc.gov/ncbddd/spanish/sicklecell/symptoms.html. Último acceso: abril de 2020.
- Chakravorty S, et al. Patient-reported experience measure in sickle cell disease. Arch Dis Child. 2018;103(12):1104-9.
- Côbo V de A, et al. Sexuality and sickle cell anemia. Rev Bras Hematol Hemoter. 2013;35(2):89-93.
- Conran N, et al. Newer aspects of the pathophysiology of sickle cell disease vaso-occlusion. Hemoglobin. 2009;33(1):1-16.
- Dampier C. Orphan drugs for sickle vaso-occlusion: Dawn of a new era of targeted treatment. Orphan Drugs: Research and Reviews 2015:5 99-112.
- de Montalembert M, et al. Transition from paediatric to adult care for patients with sickle cell disease. Br J Haematol. 2014;164(5):630-5.
- DeBaun MR, et al. Silent cerebral infarcts: a review on a prevalent and progressive cause of neurologic injury in sickle cell anemia. Blood. 2012;119(20):4587-96.
- El Khatib AM, Hayek SN. Leg ulcers in sickle cell patients: management challenges. Chronic Wound Care Management and Research. 2016;3:157-61.
- Food and Drug Administration. The Voice of the Patient. FDA report - Sickle Cell Disease. 2014. Disponible en: https://www.fda.gov/media/89898/download. Último acceso: abril de 2020.
- Hasan SP, et al. Depression in sickle cell disease. J Natl Med Assoc. 2003;95(7):533-7.
- Jenerette CM, et al. Self-Care Recommendations of Middle-Aged and Older Adults with Sickle Cell Disease. Nurs Res Pract 2011;270594.
- Key NS, Derebail VK. Sickle-cell trait: novel clinical significance. Hematology Am Soc Hematol Educ Program. 2010;2010:418-22.
- National Institutes of Health, National Heart, Lung, and Blood Institute (NHLBI), Division of Blood Diseases and Resources. The management of sickle cell disease. NIH publication No. 02-21172002. 2002.
- National Institutes of Health, National Heart, Lung, and Blood Institute (NHLBI). Sickle Cell Disease [Internet]. 2020. Disponible en: https://www.nhlbi.nih.gov/health-topics/sickle-cell-disease. Último acceso: abril de 2020.
- Piel FB, et al. Global burden of sickle cell anaemia in children under five, 2010-2050: modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013;10(7):e1001484.
- Piel FB, et al. Sickle Cell Disease. N Engl J Med. 2017;376(16):1561-73.
- Rees DC, et al. Sickle-cell disease. Lancet. 2010;376(9757):2018-31.
- Shah R, et al. Acute and chronic hepatobiliary manifestations of sickle cell disease: A review. World J Gastrointest Pathophysiol. 2017;8(3):108-16.
- Sharma S, et al. Sleep disorders in adult sickle cell patients. J Clin Sleep Med. 2015;11(3):219-23.
- Shriner D, Rotimi CN. Whole-Genome-Sequence-Based Haplotypes Reveal Single Origin of the Sickle Allele during the Holocene Wet Phase. Am J Hum Genet. 2018;102(4):547-56.
- Smith-Whitley K. Reproductive issues in sickle cell disease. Hematology Am Soc Hematol Educ Program. 2014;2014(1):418-24.
- Sociedad Española de Hematología y Oncología Pediátricas. Enfermedad de Células Falciformes - Guía de Práctica Clínica. SEHOP 2019. Disponible en: http://www.sehop.org/wp-content/uploads/2019/03/Gu%C3%ADa-SEHOP-Falciforme-2019.pdf. Último acceso: abril de 2020.
- Steinberg MH. Management of sickle cell disease. N Engl J Med. 1999;340(13):1021-30.
- Swanson ME, et al. Disability among individuals with sickle cell disease: literature review from a public health perspective. Am J Prev Med. 2011;41(6 Suppl 4):S390-7.
- U.S. National Library of Medicine. MedlinePlus. Enfermedad de células falciformes [Internet]. 2020. Disponible en: https://medlineplus.gov/spanish/sicklecelldisease.html. Último acceso: abril de 2020.
- van Tuijn CF, et al. Daily pain in adults with sickle cell disease-a different perspective. Am J Hematol. 2017;92(2):179-86.
- Webmd. What Is a Sickle Cell Crisis? [Internet]. 2019. Disponible en: https://www.webmd.com/a-to-z-guides/sickle-cell-crisis#1. Último acceso: abril de 2020.
- Zhang D, et al. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood. 2016;127(7):801-9.
- Centers for Disease Control and Prevention (CDC). National Center on Birth Defects and Developmental Disabilities, Division of Blood Disorders. Cuestionario breve sobre la enfermedad de células Falciformes [Internet]. 2013. Disponible en: https://www.cdc.gov/ncbddd/spanish/features/quiz/sicklecell/alt.html. Último acceso: abril de 2020.
- Centers for Disease Control and Prevention (CDC). National Center on Birth Defects and Developmental Disabilities, Division of Blood Disorders. Enfermedad de células falciformes - Consejos para una vida saludable. [Internet]. Disponible en: https://www.cdc.gov/ncbddd/spanish/sicklecell/documents/A_tipsheets_5_spanish.pdf. Último acceso: abril de 2020.